Annotation infrastructure
Annotation is a central step in genomic analysis. Sequencing and other genomic assays identify variants, positions, or regions, and annotation adds the biological and clinical context needed to interpret them. With annotation in place, these genomic inputs can be searched, filtered, and prioritized in a consistent and reproducible way.
GAIn is a flexible infrastructure for annotating variants, positions, and regions. Annotation is performed using a user-provided specification called an annotation pipeline.
Annotation pipelines
Annotation pipelines are YAML files with a defined structure: an optional preamble followed by an ordered list of annotators. For each annotator, the user specifies the annotator type, points to the required resources by ID, chooses the annotatable to operate on, sets any additional parameters as needed, and selects which attributes to emit in the output.
To define an annotator, you simply start by declaring the annotator type
from the available annotator types (e.g. effect_annotator, gene_score_annotator),
and then specify the settings for that annotator type as a YAML dictionary.
The exact settings depend on the annotator type. The typical structure of an annotator
declaration is shown below.
- <annotator type>
# annotator settings
Many annotators share a small set of common fields.
input_annotatable selects which annotatable object to use as input (by default,
this is the annotatable read from the input file). For gene-focused annotators, input_gene_list
specifies the gene list used to match annotatables to genes. An annotatable is the genomic
object that annotators operate on. Some annotators produce a new annotatable
(for example after liftover or allele normalization) that can be passed downstream
to later annotators.
Annotators typically specify the resources they use by fields
like resource_id, genome, and gene_models, depending on the annotator type.
Annotators typically have an attributes field which defines the attributes that will be added to the annotation by GAIn. If this is omitted then the default annotation specified in the resource is used.
The attributes section has the following minimal structure, which only specifies the source attribute in the resource:
attributes:
- source: <source attribute>
attributes section has two optional fields.
name: the user may rename the resource attribute to a different name in the annotation output. If not specified, source attribute name is used in the output.internal: the user can choose to compute an attribute but not include it in the output. This is useful when an attribute is needed as input to a later annotator but is not of interest to the user. If internal is set to true, that attribute will be computed but not written to the annotation output. The default value for internal is false, meaning that attributes are included in the output by default.
Different annotator types have different configurations, and we will discuss them below. First, we will talk about the preamble section.
Preamble
In addition to the ordered list of annotators, an annotation pipeline
config may include an optional preamble section. The preamble records
high-level information about the pipeline that is useful for human readers
and documentation tools.
A key preamble field is input_reference_genome. Many resources are reference-genome-specific,
so declaring the genome once allows annotators to reuse it without repeating the
same setting throughout the pipeline. If an annotator specifies its own genome,
that value overrides the preamble’s input_reference_genome.
When a preamble is present, annotators must be listed under the annotators key.
Below is an annotation pipeline draft that includes a preamble section.
preamble:
summary: my_summary
description: my_description
input_reference_genome: my_genome
metadata:
author: my_name
customField: "Any arbitrary key/value pairs can go here."
customNestedDictionary:
key1: value1
annotators:
- position_score_annotator:
resource_id: <position score resource ID>
attributes:
- source: <source_score_attribute>
name: <renamed_score_attribute>
Annotators
Annotators are the individual components that make up an annotation pipeline. Each annotator takes an input object (an annotatable, such as the annotatable read from the input file, or a derived annotatable produced by an earlier annotator), uses one or more resources and settings, and produces outputs in the form of annotation attributes and, in some cases, a new annotatable for downstream use. For that reason, annotators are best understood by what they consume (which annotatable or gene list they operate on) and what they produce (annotation attributes, gene context, or a transformed annotatable).
In the sections below, we group annotators by their role in the pipeline: score-based annotators, effect annotators that derive gene and transcript context, annotators that produce new annotatables for downstream annotation, gene set annotators for set membership, and plugin-based annotators such as SpliceAI.
Score annotators
Score annotators attach values from score resources to each input annotatable
and emit them as annotation attributes. In some cases, a single annotatable can match
multiple score records (for example due to overlapping intervals or multi-base events).
Aggregators define how these multiple values are combined into a single output value.
Available aggregators are mean, median, max, min, mode, join (i.e.,
join(;)), list, dict, and concatenate.
position_score_annotator
A position_score_annotator adds locus-level context to each annotatable by looking up values from a position score resource at
the annotatable’s genomic coordinates. Position score resources assign per-base metrics to fixed genomic positions independent of
the observed allele, providing signals such as evolutionary conservation or functional constraint (for example, phyloP and phastCons).
A minimal position_score_annotator configuration is shown below.
This annotator looks up the value of the source attribute from the specified position score resource and adds it to the annotation output as an attribute with the same name.
- position_score_annotator:
resource_id: <position score resource ID>
attributes:
- source: <source_score_attribute>
An annotatable may overlap multiple positions or intervals in
the underlying score resource (for example, an INDEL spans
multiple bases). In these cases, the annotator combines the
matched values using a single aggregation setting, position_aggregator.
The example below uses a position_aggregator and also renames the output attribute to renamed_score_attribute.
- position_score_annotator:
resource_id: <position score resource ID>
attributes:
- source: <source_score_attribute>
name: <renamed_score_attribute>
position_aggregator: <aggregator>
allele_score_annotator
An allele_score_annotator adds allele-level context to each annotatable by looking up values from an allele score resource for a
specific REF→ALT change (and, in some cases, local sequence context). Allele score resources capture signals such as predicted variant impact
(for example, CADD, AlphaMissense, MPC), population-level evidence like allele frequency (for example, gnomAD), and curated clinical assertions (for example, ClinVar).
A minimal allele_score_annotator configuration is shown below:
- allele_score_annotator:
resource_id: <allele score resource ID>
attributes:
- source: <source_score_attribute>
Unlike position scores, allele score lookups can involve two distinct kinds of “many-to-one”
situations: an annotatable may span multiple positions and it may also yield multiple allele-level matches. For this reason, allele_score_annotator supports two aggregation settings:
position_aggregator: combines values across multiple positions spanned by the annotatable.allele_aggregator: combines values across multiple allele-level matches for the same annotatable.
A complete attribute entry with both aggregators is shown below:
- allele_score_annotator:
resource_id: <allele score resource ID>
attributes:
- source: <source_score_attribute>
name: <renamed_score_attribute>
position_aggregator: <aggregator>
allele_aggregator: <aggregator>
gene_score_annotator
A gene_score_annotator adds gene-level context by attaching per-gene metrics to an
annotatable after it has been mapped to one or more genes. Gene score resources summarize
properties such as constraint, intolerance, and gene size, and are typically keyed by
stable gene identifiers (for example, HGNC). Common examples include pLI and LOEUF, which
reflect a gene’s intolerance to loss-of-function variation, as well as scores based on gene
length or disease association.
Unlike position and allele score annotators, a gene score annotator requires a gene
list in the annotation context. The gene list is provided via input_gene_list and is
typically produced by an upstream effect annotator (for example, gene_list or LGD_gene_list).
If the requested gene list is not present, the annotator cannot match annotatables to genes.
An example gene_score_annotator configuration is shown below:
- gene_score_annotator:
resource_id: <gene score resource ID>
input_gene_list: <gene list to use>
attributes:
- source: <source_score_attribute>
name: <renamed_score_attribute>
gene_aggregator: <aggregator>
The gene_aggregator setting controls how gene score values are combined when an annotatable
maps to multiple genes in the selected gene list.
Effect annotators
Effect annotators interpret each annotatable in the context of a gene model and report the predicted functional consequence (for example, missense, synonymous, or loss-of-function) along with the affected genes and transcripts. Unlike score-based annotators, which attach values from external resources, effect annotators derive annotation context directly from the annotatable and the gene models. Effect annotators require a gene models resource. The reference genome can be specified explicitly or inferred from the gene models configuration or the pipeline preamble.
Effect annotators also produce gene lists and effect summaries that can be consumed by
downstream gene-based annotators (for example, gene scores and gene sets via input_gene_list).
GAIn provides two effect annotators: effect_annotator, which evaluates a specific
variant change and can emit detailed transcript-level output, and simple_effect_annotator,
which uses a simplified scheme that emphasizes position-based classification.
effect_annotator
The effect_annotator predicts the functional consequence of a variant annotatable with respect to
protein-coding transcripts (for example, missense, synonymous, LGD) using the provided gene models.
It can emit both high-level summaries (such as the worst effect) and more detailed per-gene and
per-transcript outputs.
A minimal effect_annotator configuration is shown below:
- effect_annotator:
gene_models: <gene models resource ID>
genome: <reference genome resource ID>
The genome field is optional. If it is not provided, the annotator resolves the reference genome in the following order:
1. genome specified in the annotator configuration2. reference_genome label in the configured gene_models resource3. input_reference_genome from the pipeline preamble
The effect_annotator can emit the following attributes:
worst_effect (default: yes): the worst effect across all transcripts.worst_effect_genes (default: yes): comma-separated list of genes with the worst effect.gene_effects (default: yes): effect types for each gene.effect_details (default: yes): effect details for each affected transcript.gene_list (internal, default: yes): list of all affected genes.worst_effect_gene_list (internal, default: no): list of genes with the worst effect.genes (default: no): comma-separated list of affected genes.<effect>_genes (default: no): comma-separated list of genes with a specific effect type.<effect>_gene_list (internal, default: no): list of genes with a specific effect type.
The effect_annotator example below uses the MANE 1.5 gene models in the IossifovLab GRR.
Since this gene models resource already specifies its reference genome via its configuration labels,
the genome field is not required in the annotator configuration.
The example also renames worst_effect to MANE_1.5_worst_effect.
- effect_annotator:
gene_models: hg38/gene_models/MANE/1.5
attributes:
- source: worst_effect
name: MANE_1.5_worst_effect
simple_effect_annotator
The simple_effect_annotator assigns a coarse, position-based classification to each annotatable
using the provided gene models. Conceptually, it first separates loci into broad
categories such as intergenic vs genic, and then refines genic loci into coding and several
noncoding classes (as in the scheme shown below, reproduced from PMID: 34471188). Unlike
effect_annotator, which evaluates the specific REF→ALT change, the simple effect annotator
emphasizes where the locus falls relative to gene structure.
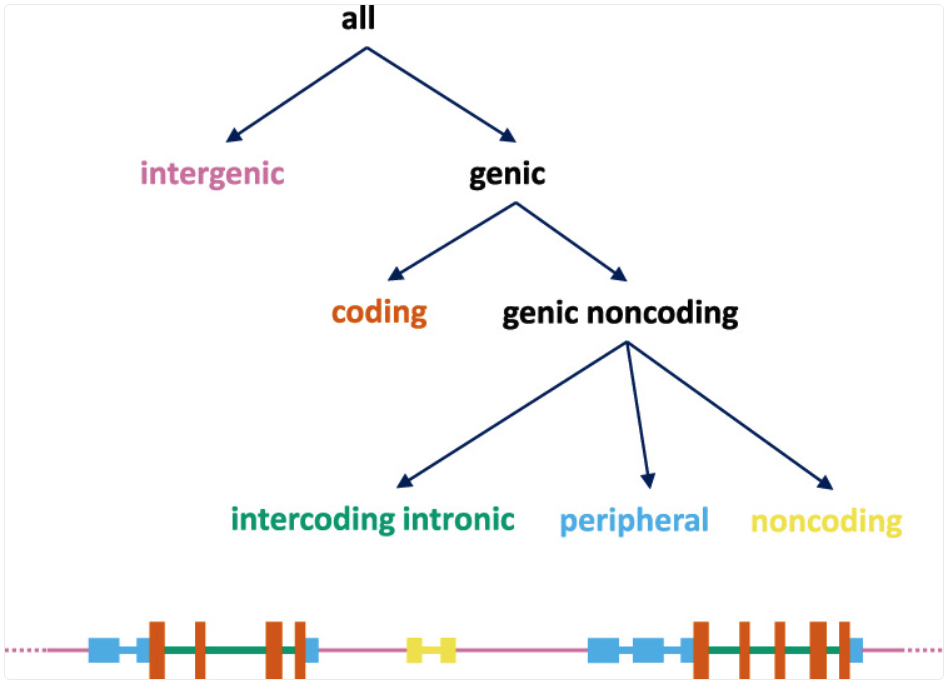
Event types defined by the simple_effect_annotator.
A minimal simple_effect_annotator configuration is shown below:
- simple_effect_annotator:
gene_models: <gene models resource ID>
The output fields follow the same general pattern as effect_annotator (for example,
a “worst” class plus affected gene lists), but the effect labels reflect this simplified,
location-focused scheme.
Transforming annotators
Transforming annotators produce a new annotatable derived from the input, rather than
adding annotation attributes. They take an annotatable,
apply a transformation such as liftover, allele normalization, or chromosome renaming, and emit
a new annotatable that can be passed downstream to later annotators via input_annotatable.
These annotators are commonly used to reconcile differences in reference genome, coordinate
conventions, or chromosome naming before running additional annotation steps.
liftover_annotator
The liftover_annotator maps an annotatable from one reference genome to another using a liftover chain.
Its primary output is a new annotatable in the target reference genome. By default,
this annotatable is named liftover_annotatable and can be passed to downstream annotators via
input_annotatable.
The attributes section is used to rename the produced annotatable (and optionally mark
it as internal), rather than to select score fields from a resource. A typical configuration
is shown below. Here, the lifted-over annotatable is renamed to T2T_annotatable and marked as
internal so it can be used downstream without appearing in the final output.
- liftover_annotator:
chain: liftover/hg38_to_T2T
source_genome: hg38/genomes/GRCh38.p14
target_genome: t2t/genomes/t2t-chm13v2.0
attributes:
- source: liftover_annotatable
name: T2T_annotatable
internal: true
The source_genome and target_genome fields are optional. If they are not provided,
the annotator attempts to infer them from the liftover chain resource configuration (via
source_genome and target_genome labels in the chain resource’s meta section).
normalize_allele_annotator
The normalize_allele_annotator converts a variant annotatable to a canonical allele representation using
the normalization algorithm described here.
It produces a new annotatable named normalized_allele, which can be passed downstream via
input_annotatable. This is commonly used before running ``allele_score_annotator``s, to ensure lookups match the allele representation used by the underlying resources.
As with liftover, the attributes section is used to rename the produced annotatable
(and optionally mark it as internal). A typical configuration is shown below:
- normalize_allele_annotator:
genome: hg38/genomes/GRCh38-hg38
attributes:
- source: normalized_allele
name: hg38_normalized_annotatable
internal: true
This annotator does not require a resource and can be used with no specifications as follows.
In this case, the output annotatable is named normalized_allele and is marked as internal,
so it is not included in the annotation output.
- normalize_allele_annotator
chrom_mapping
The chrom_mapping annotator rewrites chromosome (contig) names to match a different
naming convention. This is useful when a score resource, gene models, or an input file uses
contig names that do not match the reference genome (for example, with or without a chr prefix,
or with assembly-specific contig labels).
The annotator produces a new annotatable named renamed_chromosome, which can be passed
downstream via input_annotatable. By default, this output is treated as an internal attribute,
since it is primarily used as an intermediate annotatable rather than a final annotation field.
The annotator supports two common modes: prefix rewriting and explicit name mapping. This example removes the chr prefix from contig names (for example, chr1 → 1).
- chrom_mapping:
del_prefix: chr
attributes:
- source: renamed_chromosome
internal: true
This example rewrites specific contig names using an explicit mapping table (for example, 1 → chr1 and MT → chrM). This is useful when the naming differences are not just a simple prefix change.
- chrom_mapping:
mapping:
"1": chr1
"2": chr2
"X": chrX
"Y": chrY
"MT": chrM
cnv_collection_annotator
cnv_collection annotator reports copy-number variants (CNVs) that overlap each input locus.
A CNV collection resource stores CNV intervals (and optional associated fields such as CNV type, frequency, or dataset labels),
and during annotation GAIn performs an interval overlap query at the annotatable’s genomic coordinates to retrieve matching CNV events.
A cnv_collection_annotator can be used with a minimal configuration. If you omit the attributes section, GAIn uses the resource’s default annotation,
which reports the count of overlapping CNV events in the collection for each input annotatable (i.e., how many CNVs in the database overlap that locus).
- cnv_collection_annotator:
resource_id: <resource id>
When you do specify attributes, the syntax is slightly different from score annotators:
you request fields using the attribute.<id> form, where <id> refers to an exposed attribute
in the CNV collection resource (for example, class, frequency, dataset label). In addition, you can use
cnv_filter: and request only results gated by specific values of an attribute.
- cnv_collection_annotator:
resource_id: <resource id>
cnv_filter: <attribute1 id> == deletion
attributes:
- attribute.<attribute1 id>
- attribute.<attribute2 id>
gene_set_annotator
The gene_set_annotator reports whether genes implicated by an annotatable belong to gene sets from a
gene set collection. Gene set resources group genes by shared functions, pathways, or phenotypes
(for example, GO categories or MSigDB collections). In an annotation pipeline, the gene set annotator
operates on a gene list in the annotation context (provided via input_gene_list) and emits membership
information as annotation attributes.
The annotator can emit a membership attribute for each gene set in the collection and/or a
special attribute in_sets, which is the list of gene set names that the annotatable’s genes belong
to (based on the selected input gene list). A typical configuration is shown below:
- gene_set_annotator:
resource_id: <gene set collection resource ID>
input_gene_list: <gene list produced by matching annotatables to gene models>
attributes:
- <gene set name>
- in_sets
spliceai_annotator
The spliceai_annotator plugin is a wrapper around SpliceAI, which predicts the impact of variants on splicing. It annotates variants with delta scores (DS) and delta positions (DP) for acceptor gain/loss and donor gain/loss, along with supporting fields such as affected gene symbol and transcript IDs.
To install the plugin, run:
mamba install \
-c conda-forge \
-c bioconda \
-c iossifovlab \
gpf_spliceai_annotator
A typical configuration is shown below:
- spliceai_annotator:
genome: hg38/genomes/GRCh38-hg38
gene_models: hg38/gene_models/refSeq_v20200330
distance: 50
mask: false
The configuration fields are:
genome: the reference genome resource ID to use for the annotation.gene_models: the gene models resource ID to use for the annotation.distance: maximum distance (in bp) between the variant and a gained/lost splice site (default: 50).mask: if set to true, masks scores representing annotated acceptor/donor gain and unannotated acceptor/donor loss (default: false).
The annotator produces the following attributes:
gene: gene symbol.transcript_ids: comma-separated list of transcript IDs.DS_AG: delta score for acceptor gain.DS_AL: delta score for acceptor loss.DS_DG: delta score for donor gain.DS_DL: delta score for donor loss.DS_MAX: maximum delta score.DP_AG: delta position for acceptor gain.DP_AL: delta position for acceptor loss.DP_DG: delta position for donor gain.DP_DL: delta position for donor loss.ref_A_p: reference acceptor probabilities.ref_D_p: reference donor probabilities.alt_A_p: alternative acceptor probabilities.alt_D_p: alternative donor probabilities.delta_score: compact SpliceAI annotation string with DS and DP values. Format:ALLELE|SYMBOL|DS_AG|DS_AL|DS_DG|DS_DL|DP_AG|DP_AL|DP_DG|DP_DL.
VEP annotators
GAIn provides two annotators that run the Ensembl Variant Effect Predictor (VEP)
via Docker to produce VEP-based consequence annotations. Both annotators are designed
to be run in batch mode and expose VEP outputs as annotation attributes. The two annotators
differ primarily in how VEP is configured: vep_full_annotator uses a local VEP cache,
while external_vep_gtf_annotator runs VEP against GAIn-provided genome and gene models
from a GRR.
Use the VEP Full Annotator when you have (or want) a local VEP cache and need access to the broadest set of VEP output fields. Use the VEP Effect Annotator when you want VEP to run against the genome and gene models already available in your GRR (for example MANE), so the results align with the resources used elsewhere in the pipeline.
Using the VEP annotators requires the gpf_vep_annotator conda package and a working Docker installation.
mamba install \
-c conda-forge \
-c bioconda \
-c iossifovlab \
-c defaults \
gpf_vep_annotator
The VEP annotators can be run only in batch mode.
vep_full_annotator
The vep_full_annotator runs Ensembl VEP using a local VEP cache directory, allowing access to the broadest set of VEP output fields. This annotator is typically used when you want standard VEP annotations directly from the Ensembl cache and plan to select a subset of VEP fields as pipeline attributes.
The full VEP annotator requires a VEP cache to be accessible on the local file system. You can download the cache using the vep_install tool provided by the ensembl-vep conda package (installed as part of gpf_vep_annotator). For example, to download the cache for hg38:
vep_install -a cf -s homo_sapiens -y GRCh38 -c /output/path/to/cache --convert
The annotator configuration looks like this:
- vep_full_annotator:
cache_dir: <VEP cache directory>
vep_version: <VEP version to use>
cache_dir: path to the VEP cache directory (the same directory passed as -c to vep_install).vep_version: VEP version to use. If not specified, the annotator uses the latest available Docker image from ensemblorg/ensembl-vep. Versions may be given with or without a minor version (for example, 113, 113.0, 113.3).
By default, the annotator produces only the following attributes: SYMBOL, Feature, Feature_type, Consequence, worst_consequence, highest_impact, and gene_consequence.
The full VEP annotator can optionally emit many additional VEP fields (listed below) by selecting them as pipeline attributes. VEP annotators are run via annotate_columns in batch mode. See the example command at the end of the VEP Effect Annotator section.
All available output attributes:
Gene: Stable ID of affected geneFeature: Stable ID of featureFeature_type: Type of feature - Transcript, RegulatoryFeature or MotifFeatureConsequence: Consequence typecDNA_position: Relative position of base pair in cDNA sequenceCDS_position: Relative position of base pair in coding sequenceProtein_position: Relative position of amino acid in proteinAmino_acids: Reference and variant amino acidsCodons: Reference and variant codon sequenceExisting_variation: Identifier(s) of co-located known variantsIMPACT: Subjective impact classification of consequence typeDISTANCE: Shortest distance from variant to transcriptSTRAND: Strand of the feature (1/-1)FLAGS: Transcript quality flagsVARIANT_CLASS: SO variant classSYMBOL: Gene symbol (e.g. HGNC)SYMBOL_SOURCE: Source of gene symbolHGNC_ID: Stable identifier of HGNC gene symbolBIOTYPE: Biotype of transcript or regulatory featureCANONICAL: Indicates if transcript is canonical for this geneMANE: MANE (Matched Annotation from NCBI and EMBL-EBI) set(s) the transcript belongs toMANE_SELECT: MANE Select (Matched Annotation from NCBI and EMBL-EBI) TranscriptMANE_PLUS_CLINICAL: MANE Plus Clinical (Matched Annotation from NCBI and EMBL-EBI) TranscriptTSL: Transcript support levelAPPRIS: Annotates alternatively spliced transcripts as primary or alternate based on a range of computational methodsCCDS: Indicates if transcript is a CCDS transcriptENSP: Protein identiferSWISSPROT: UniProtKB/Swiss-Prot accessionTREMBL: UniProtKB/TrEMBL accessionUNIPARC: UniParc accessionUNIPROT_ISOFORM: Direct mappings to UniProtKB isoformsGENE_PHENO: Indicates if gene is associated with a phenotype, disease or traitSIFT: SIFT prediction and/or scorePolyPhen: PolyPhen prediction and/or scoreEXON: Exon number(s) / totalINTRON: Intron number(s) / totalDOMAINS: The source and identifer of any overlapping protein domainsmiRNA: SO terms of overlapped miRNA secondary structure feature(s)HGVSc: HGVS coding sequence nameHGVSp: HGVS protein sequence nameHGVS_OFFSET: Indicates by how many bases the HGVS notations for this variant have been shiftedAF: Frequency of existing variant in 1000 Genomes combined populationAFR_AF: Frequency of existing variant in 1000 Genomes combined African populationAMR_AF: Frequency of existing variant in 1000 Genomes combined American populationEAS_AF: Frequency of existing variant in 1000 Genomes combined East Asian populationEUR_AF: Frequency of existing variant in 1000 Genomes combined European populationSAS_AF: Frequency of existing variant in 1000 Genomes combined South Asian populationgnomADe_AF: Frequency of existing variant in gnomAD exomes combined populationgnomADe_AFR_AF: Frequency of existing variant in gnomAD exomes African/American populationgnomADe_AMR_AF: Frequency of existing variant in gnomAD exomes American populationgnomADe_ASJ_AF: Frequency of existing variant in gnomAD exomes Ashkenazi Jewish populationgnomADe_EAS_AF: Frequency of existing variant in gnomAD exomes East Asian populationgnomADe_FIN_AF: Frequency of existing variant in gnomAD exomes Finnish populationgnomADe_MID_AF: Frequency of existing variant in gnomAD exomes Mid-eastern populationgnomADe_NFE_AF: Frequency of existing variant in gnomAD exomes Non-Finnish European populationgnomADe_OTH_AF: Frequency of existing variant in gnomAD exomes other combined populationsgnomADe_SAS_AF: Frequency of existing variant in gnomAD exomes South Asian populationgnomADe_REMAINING_AF: Frequency of existing variant in gnomAD exomes remaining combined populationsgnomADg_AF: Frequency of existing variant in gnomAD genomes combined populationgnomADg_AFR_AF: Frequency of existing variant in gnomAD genomes African/American populationgnomADg_AMI_AF: Frequency of existing variant in gnomAD genomes Amish populationgnomADg_AMR_AF: Frequency of existing variant in gnomAD genomes American populationgnomADg_ASJ_AF: Frequency of existing variant in gnomAD genomes Ashkenazi Jewish populationgnomADg_EAS_AF: Frequency of existing variant in gnomAD genomes East Asian populationgnomADg_FIN_AF: Frequency of existing variant in gnomAD genomes Finnish populationgnomADg_MID_AF: Frequency of existing variant in gnomAD genomes Mid-eastern populationgnomADg_NFE_AF: Frequency of existing variant in gnomAD genomes Non-Finnish European populationgnomADg_OTH_AF: Frequency of existing variant in gnomAD genomes other combined populationsgnomADg_SAS_AF: Frequency of existing variant in gnomAD genomes South Asian populationgnomADg_REMAINING_AF: Frequency of existing variant in gnomAD genomes remaining combined populationsMAX_AF: Maximum observed allele frequency in 1000 Genomes, ESP and ExAC/gnomADMAX_AF_POPS: Populations in which maximum allele frequency was observedCLIN_SIG: ClinVar clinical significance of the dbSNP variantSOMATIC: Somatic status of existing variantPHENO: Indicates if existing variant(s) is associated with a phenotype, disease or trait; multiple values correspond to multiple variantsPUBMED: Pubmed ID(s) of publications that cite existing variantMOTIF_NAME: The stable identifier of a transcription factor binding profile (TFBP) aligned at this positionMOTIF_POS: The relative position of the variation in the aligned TFBPHIGH_INF_POS: A flag indicating if the variant falls in a high information position of the TFBPMOTIF_SCORE_CHANGE: The difference in motif score of the reference and variant sequences for the TFBPTRANSCRIPTION_FACTORS: List of transcription factors which bind to the transcription factor binding profileworst_consequence: Worst consequence reported by VEPhighest_impact: Highest impact reported by VEPgene_consequence: List of gene consequence pairs reported by VEP
external_vep_gtf_annotator
The external_vep_gtf_annotator (VEP Effect Annotator) runs Ensembl VEP via Docker using the genome and gene models available in your GRR. This is useful when you want VEP consequences and related fields while keeping the annotation aligned with the same genome and gene models used elsewhere in GAIn pipelines. The VEP annotators can be run only in batch mode.
The annotator configuration looks like this:
- external_vep_gtf_annotator:
genome: hg38/genomes/GRCh38-hg38
gene_models: hg38/gene_models/MANE/1.5
vep_version: <VEP version to use>
genome: the reference genome resource ID to use for the annotation.gene_models: the gene models resource ID to use for the annotation.vep_version: VEP version to use. If not specified, the annotator uses the latest available Docker image from ensemblorg/ensembl-vep. Versions may be given with or without a minor version (for example, 113, 113.0, 113.3). If only the major version is provided (e.g., 113), it is interpreted as 113.0.
By default, only the following are produced: SYMBOL, Feature, Feature_type, Consequence, worst_consequence, highest_impact, gene_consequence,
and the value from the provided gene models.
The VEP effect annotator can optionally emit additional VEP fields (listed below) by selecting them as pipeline attributes.
All available output attributes:
Location: Location of variant in standard coordinate format (chr:start or chr:start-end)Allele: The variant allele used to calculate the consequenceGene: Stable ID of affected geneFeature: Stable ID of featureFeature_type: Type of feature - Transcript, RegulatoryFeature or MotifFeatureConsequence: Consequence typecDNA_position: Relative position of base pair in cDNA sequenceCDS_position: Relative position of base pair in coding sequenceProtein_position: Relative position of amino acid in proteinAmino_acids: Reference and variant amino acidsCodons: Reference and variant codon sequenceExisting_variation: Identifier(s) of co-located known variantsIMPACT: Subjective impact classification of consequence typeDISTANCE: Shortest distance from variant to transcriptSTRAND: Strand of the feature (1/-1)FLAGS: Transcript quality flagsSYMBOL: Gene symbol (e.g. HGNC)SYMBOL_SOURCE: Source of gene symbolHGNC_ID: Stable identifer of HGNC gene symbolSOURCE: Source of transcriptworst_consequence: Worst consequence reported by VEPhighest_impact: Highest impact reported by VEPgene_consequence: List of gene consequence pairs reported by VEP<gene model filename>: Value from provided gene models
With a prepared variants file and an annotation.yaml pipeline configuration, VEP-based annotation can be run via annotate_columns in batch mode using the –batch-mode flag. For example:
annotate_columns ./variants.tsv.gz ./annotation.yaml \
-w work -o ./out.tsv -v -j 4 --batch-mode \
--col-chrom CHROM --col-pos POS --col-ref REF -r 10000 --col-alt ALT \
--allow-repeated-attributes
Command line tools
GAIn provides three command-line tools for working with annotation pipelines. Two tools run annotation over different input formats (tabular files or VCF). A third tool generates a human-readable HTML description of a pipeline for documentation and review.
annotate_columns: annotate delimiter-separated tabular files (TSV/CSV and similar)
annotate_vcf: annotate VCF/VCF.gz files
annotate_doc: render a pipeline YAML into a readable HTML document
Notes on usage
Across the two annotation runners (annotate_columns, annotate_vcf), the same basic pattern applies:
Input data: the dataset to be annotated.
Pipeline configuration: a pipeline YAML file that defines the ordered list of annotators (and an optional preamble).
Output: an annotated dataset that includes the requested attributes.
Parallel execution: tools parallelize their workload when possible and will attempt to do so by default. Parallel runs create task status flags/logs and may create a work directory for intermediate outputs. If a re-run appears to skip tasks due to existing task state, remove the task-status directory (and any tool-specific work directory) so tasks can be executed again.
Re-annotation: use
--reannotatewhen you want to update or recompute only part of an already annotated dataset, and--full-reannotationto ignore prior results and recompute everything.Repeated attributes: use
--allow-repeated-attributes(short form -ar) to allow duplicate attribute names. Repeated fields are disambiguated by appending the annotator ID (e.g.,_A0,_A1) to the attribute name.Capture logs for reproducibility: for long runs, prefer –logfile and keep the pipeline YAML alongside the produced output so the annotation can be reproduced later.
Indexing improves parallelization: for bgzip-compressed tabular and VCF files, tabix-indexing enables region-based task splitting (via
annotate_columnsandannotate_vcf), which is typically faster and more memory efficient than single-process runs.Tabular inputs (annotate_columns): Be explicit about annotatable columns when needed:
annotate_columnstries to infer the chromosome/position/ref/alt columns from the header, but for nonstandard headers you should pass the appropriate--col-*arguments to avoid mis-detection.
annotate_columns
annotate_columns annotates delimiter-separated tabular files (TSV, CSV, and similar).
It reads annotatables from columns in the input table, runs the specified annotation pipeline,
and writes an annotated table as output. annotate_columns works for all annotatables (variant, position, region).
The minimal invocation is the input table and the pipeline YAML:
annotate_columns input.tsv annotation.yaml
By default, the output is written next to the input as input_annotated.tsv. To choose a different output filename, use -o / –output:
annotate_columns input.tsv annotation.yaml -o my_output.tsv
The input file should be a table with a header. annotate_columns identifies annotatable fields from column names when
possible (e.g., chromosome, position, reference, alternative, or interval columns). If your input uses nonstandard names, map them explicitly with --col-* options.
For example, if the chromosome column is named CHROMOSOME (instead of the default chrom), you can run:
annotate_columns input.tsv annotation.yaml --col-chrom CHROMOSOME
Common column mapping flags are:
--col-chrom
--col-pos
--col-ref
--col-alt
Additional patterns (such as --col-pos-beg / --col-pos-end, --col-location, or --col-variant) can be used when your input encodes annotatables in alternative representations.
Common options:
-o, –output: output filename.
-w, –work-dir: directory for intermediate files.
-j, –jobs: number of parallel jobs.
-r, –region-size: region size used for splitting tabix-indexed inputs.
–input-separator / –output-separator: override input/output delimiters.
–reannotate and –full-reannotation: re-run annotation over existing outputs.
-ar, –allow-repeated-attributes: allow duplicate attribute names by suffixing them with annotator IDs.
For a full list of options run annotate_columns --help
annotate_vcf
annotate_vcf annotates variants in VCF (or bgzip-compressed *.vcf.gz) files.
It reads each VCF record as the input annotatable, runs the specified annotation pipeline,
and writes an annotated VCF as output.
The minimal invocation is the input VCF and the pipeline YAML:
annotate_vcf input.vcf.gz annotation.yaml
By default, the output is written next to the input as input_annotated.vcf.gz.
To choose a different output filename, use -o or --output:
annotate_vcf input.vcf.gz annotation.yaml -o my_output.vcf.gz
If the file is tabix-indexed, annotate_vcf can split the work by genomic region for parallel execution.
Common options:
-o, –output: output filename.
-w, –work-dir: directory for intermediate files.
-j, –jobs: number of parallel jobs.
-r, –region-size: region size used for splitting tabix-indexed inputs.
–reannotate and –full-reannotation: re-run annotation over existing outputs.
-ar, –allow-repeated-attributes: allow duplicate attribute names by suffixing them with annotator IDs.
For a full list of options run annotate_vcf --help
annotate_doc
annotate_doc generates a human-readable HTML document from an annotation pipeline YAML.
This is useful for reviewing a pipeline, sharing it with collaborators, or publishing a readable
description alongside your analysis.
Partial screen shot of the summary html page created for T2T annotation pipeline in IossifovLab GRR.
The minimal invocation is the pipeline YAML:
annotate_doc annotation.yaml
By default, the tool writes an HTML file next to the pipeline (using its default naming). To choose the output filename, use -o or --output:
annotate_doc annotation.yaml -o annotation.html
Common options:
-o, –output: output HTML filename.
–verbose: increase logging.
–logfile: write logs to a file.
-i, –instance / -g, –grr / –grr-directory: control which GRR context is used when resolving pipeline resources.
For a full list of options run annotate_doc --help
Example annotations
1: Effect annotation
Let’s revisit the three variants we annotated in the Getting started on the CLI section. In this section, we will walk through example annotation pipelines for these variants. All annotations will use resources from the public IossifovLab GRR.
chrom |
pos |
ref |
alt |
|---|---|---|---|
chr14 |
21415880 |
G |
A |
chr17 |
7674904 |
TCT |
T |
chr7 |
117587806 |
G |
A |
The input consists of chromosomal positions, the reference allele, and the alternate allele. Create a file named variants.txt with this content in a working folder of your choice.
Our first example annotation pipeline includes a preamble section with high-level metadata and input_reference_genome,
which is used by annotators below unless an annotator explicitly specifies its own genome. Create a file
named annotation_pipeline_1.yaml with the following content:
preamble:
summary: Demo pipeline
description: Demonstrates a GAIn pipeline
input_reference_genome: hg38/genomes/GRCh38-hg38
annotators:
- effect_annotator:
gene_models: hg38/gene_models/MANE/1.5
attributes:
- source: genes
name: MANE_1.5_genes
- source: worst_effect
Since there is a preamble section, annotators must be specified under the annotators section. effect_annotator will use the MANE 1.5 gene models and
GRCh38-hg38 as its reference genome (as set by the preamble).
The attributes added will be genes (affected genes) and worst_effect across transcripts.
The genes attribute is renamed to MANE_1.5_genes in the output. (Because worst_effect is not renamed, it will appear as worst_effect in the output.)
To run this annotation pipeline, enter the following command, which annotates variants.txt using the pipeline in annotation_pipeline_1.yaml:
annotate_columns variants.txt annotation_pipeline_1.yaml
After the run is complete, there will be a new file called variants_annotated.txt, which includes
two additional columns showing the genes affected by each variant and the corresponding worst effect.
chrom |
pos |
ref |
alt |
MANE_1.5_genes |
worst_effect |
|---|---|---|---|---|---|
chr14 |
21415880 |
G |
A |
CHD8 |
nonsense |
chr17 |
7674904 |
TCT |
T |
TP53 |
frame-shift |
chr7 |
117587806 |
G |
A |
CFTR |
missense |
2: Position score annotation
In our next example, let’s use a minimal annotation pipeline that consists of a single position score annotator using phyloP7way.
Position scores are allele-independent and do not require allele normalization; they simply look up values at the input coordinates.
It is up to the user to ensure that the input annotatables and the position score resource are on the same assembly (for example, hg38).
position_score_annotator:
resource_id: hg38/scores/phyloP7way
Create a file called annotation_pipeline_2.yaml with this content. This time, specify the output filename to avoid overwriting the annotations from the previous example.
annotate_columns variants.txt annotation_pipeline_2.yaml -o annotation_2.txt
After the run is complete, a new file called annotation_2.txt will appear with the content below. The phyloP7way column indicates the evolutionary conservation at these genomic positions.
chrom |
pos |
ref |
alt |
phyloP7way |
|---|---|---|---|---|
chr14 |
21415880 |
G |
A |
0.917 |
chr17 |
7674904 |
TCT |
T |
-0.12 |
chr7 |
117587806 |
G |
A |
0.917 |
We note that this minimal example did not specify annotation attributes. As a result, the resource’s default_annotation is used, which (for this resource) selects a single score, phyloP7way.
In terms of output, the following pipeline produces the same result.
position_score_annotator:
resource_id: hg38/scores/phyloP7way
attributes:
- source: phyloP7way
When an attribute is not renamed, it can be written without the keyword source. The following is also an equivalent annotation pipeline.
position_score_annotator:
resource_id: hg38/scores/phyloP7way
attributes:
- phyloP7way
If you want to use the default annotation attributes and configurations of a resource, you can also use the short-hand definition shown below.
Since phyloP7way has a single score, its default_annotation is phyloP7way. Therefore, the following is also an equivalent annotation pipeline.
position_score_annotator: hg38/scores/phyloP7way
3: Allele score annotation
To annotate with allele scores, GAIn typically requires alleles to be
normalized first using normalize_allele_annotator. This annotator converts input variants to a canonical allele representation and produces a normalized_allele annotatable that
downstream allele_score_annotators can use for reliable lookups.
Allele score resources are assembly-specific. The allele_score_annotator does not perform assembly conversion, so you must ensure that the input variants and the allele score resource
correspond to the same reference genome. In this example, we declare the input genome in the pipeline preamble.
Create a file called annotation_pipeline_3.yaml with the following content. The pipeline first produces a normalized allele and then queries ClinVar for two ClinVar fields,
CLNSIG and CLNDN, renaming them in the output for readability.
preamble:
input_reference_genome: hg38/genomes/GRCh38-hg38
annotators:
- normalize_allele_annotator # >----->----->----->---┐
# |
- allele_score_annotator: # |
resource_id: hg38/scores/ClinVar_20251019 # |
input_annotatable: normalized_allele # <-----<-----┘
attributes:
- source: CLNSIG
name: clinical significance
- source: CLNDN
name: disease name
To annotate with this pipeline, run:
annotate_columns variants.txt annotation_pipeline_3.yaml -o annotation_3.txt
This produces annotation_3.txt with the requested ClinVar fields:
chrom |
pos |
ref |
alt |
clinical significance |
disease name |
|---|---|---|---|---|---|
chr14 |
21415880 |
G |
A |
Pathogenic/Likely_pathogenic |
not_provided|Intellectual_developmental_disorder_with_autism_and_macrocephaly |
chr17 |
7674904 |
TCT |
T |
Pathogenic |
Hereditary_cancer-predisposing_syndrome|TP53-related_disorder|not_provided|Li-Fraumeni_syndrome_1|Ovarian_neoplasm|Li-Fraumeni_syndrome |
chr7 |
117587806 |
G |
A |
Pathogenic |
CFTR-related_disorder|Cystic_fibrosis|Congenital_bilateral_aplasia_of_vas_deferens_from_CFTR_mutation|not_provided|Hereditary_pancreatitis|Bronchiectasis_with_or_without_elevated_sweat_chloride_1|ivacaftor_response_-_Efficacy |
4: Gene score annotation
Gene score resources provide per-gene metrics (for example constraint or intolerance scores). Unlike position- or allele-based resources, gene scores are keyed by gene identifiers, so a gene score annotator needs a gene list rather than a raw annotatable as input.
Because the input to an annotation run is an annotatable, the pipeline must
first map each annotatable to one or more genes using an effect annotator. The effect annotator can
emit one or more gene-list attributes (e.g., gene_list). These are Python lists of gene
symbols/IDs which can then be passed downstream to gene-based annotators via the input_gene_list setting.
In this example, we use the effect annotator to produce a gene_list attribute and then annotate
those genes with the pLI gene score (probability of loss-of-function intolerance).
Create a file called annotation_pipeline_4.yaml with the following content:
annotators:
- effect_annotator:
gene_models: hg38/gene_models/MANE/1.5
genome: hg38/genomes/GRCh38-hg38
attributes:
- genes
- worst_effect
- source: gene_list # >-------->--------┐
internal: false # |
# |
- gene_score_annotator: # |
resource_id: gene_properties/gene_scores/pLI # |
input_gene_list: gene_list # <--------<--------┘
attributes:
- pLI
In this pipeline, we explicitly include gene_list in the output by setting internal: false.
Otherwise, gene_list can be kept as an internal intermediate attribute that is available for downstream
annotators but not written to the final output. To annotate with this pipeline, run:
annotate_columns variants.txt annotation_pipeline_4.yaml -o annotation_4.txt
This produces annotation_4.txt with the effect outputs plus the requested gene score:
chrom |
pos |
ref |
alt |
genes |
worst_effect |
gene_list |
pLI |
|---|---|---|---|---|---|---|---|
chr14 |
21415880 |
G |
A |
CHD8 |
nonsense |
[‘CHD8’] |
{‘CHD8’: 1.0} |
chr17 |
7674904 |
TCT |
T |
TP53 |
frame-shift |
[‘TP53’] |
{‘TP53’: 0.9122229533} |
chr7 |
117587806 |
G |
A |
CFTR |
missense |
[‘CFTR’] |
{‘CFTR’: 2.96e-36} |
Gene score outputs are dictionaries mapping each matched gene to its score (for example, {‘TP53’: 0.912…}), to support annotatables with multiple gene matches.
5: Liftover annotation
Sometimes the resources you want to use are available only for a different reference genome build.
In that case, you can use liftover_annotator to convert the input annotatable to a different genome
and expose the lifted-over coordinates as a new annotatable that downstream annotators can consume.
In this example, we start from hg38 annotatables, lift them over to hg19, and then query
an hg19-only position score resource (FitCons i6 merged). The liftover_annotator emits a
built-in liftover_annotatable. We rename it to hg19_annotatable and set internal: false so it is
included in the output table. source_genome and target_genome are shown here for clarity. When the selected
chain resource already encodes the source and target genomes (as labels/metadata), these fields are typically optional.
Create a file called annotation_pipeline_6.yaml with the following content:
annotators:
- liftover_annotator:
chain: liftover/hg38_to_hg19
source_genome: hg38/genomes/GRCh38-hg38
target_genome: hg19/genomes/GATK_ResourceBundle_5777_b37_phiX174
attributes:
- source: liftover_annotatable
name: hg19_annotatable # >----->----->----->----->-------->---┐
internal: false # |
# |
- position_score_annotator: # |
resource_id: hg19/scores/FitCons-i6-merged # |
input_annotatable: hg19_annotatable # <-----<-----<-----<-----<-----<------┘
To annotate with this pipeline, run:
annotate_columns variants.txt annotation_pipeline_6.yaml -o annotation_6.txt
This produces annotation_6.txt with the lifted-over annotatable plus the hg19 score:
chrom |
pos |
ref |
alt |
hg19_annotatable |
fitcons_i6_merged |
|---|---|---|---|---|---|
chr14 |
21415880 |
G |
A |
VCFAllele(14,21884039,G,A) |
0.707 |
chr17 |
7674904 |
TCT |
T |
VCFAllele(17,7578221,TTC,T) |
0.722 |
chr7 |
117587806 |
G |
A |
VCFAllele(7,117227860,G,A) |
0.554 |
6. CNV collection annotation
A cnv_collection_annotator reports copy-number variant (CNV) events whose intervals overlap each
input locus. If you do not specify any attributes, the annotator reports the number of overlapping
CNV events observed in the collection as count.
Create a file called annotation_pipeline_cnv_1.yaml with the following content. This will query the DGV resource.
- cnv_collection_annotator:
resource_id: hg38/cnv_collections/DGV
To run this pipeline, execute:
annotate_columns variants.txt annotation_pipeline_cnv_1.yaml -o annotation_cnv_1.txt
This produces annotation_cnv_1.txt with the default count column:
chrom |
pos |
ref |
alt |
count |
|---|---|---|---|---|
chr14 |
21415880 |
G |
A |
0 |
chr17 |
7674904 |
TCT |
T |
2 |
chr7 |
117587806 |
G |
A |
2 |
To emit specific fields from the CNV records, add an attributes list. CNV collection attributes
use the attribute.<id> form (for example, attribute.deletion_duplication).
Create a file called annotation_pipeline_cnv_2.yaml:
- cnv_collection_annotator:
resource_id: hg38/cnv_collections/DGV
attributes:
- attribute.deletion_duplication
- attribute.cnv_name
Run:
annotate_columns variants.txt annotation_pipeline_cnv_2.yaml -o annotation_cnv_2.txt
This produces annotation_cnv_2.txt with the requested event-level fields:
chrom |
pos |
ref |
alt |
attribute.deletion_duplication |
attribute.cnv_name |
|---|---|---|---|---|---|
chr14 |
21415880 |
G |
A |
||
chr17 |
7674904 |
TCT |
T |
lossloss |
nsv574322nsv457659 |
chr7 |
117587806 |
G |
A |
inversionloss |
nsv7405esv3891197 |
7: Gene set annotation
Gene set resources group genes into named sets (for example pathways, functions, or phenotypes).
The gene_set_annotator can (1) report membership for specific sets you select and (2)
emit a combined list of all sets a gene belongs to.
Because gene-set membership is defined per gene (not per raw annotatable), the pipeline must first
map each annotatable to one or more genes. Here we use an effect_annotator to produce a gene_list
attribute that is then passed into the gene_set_annotator via input_gene_list.
In this example, we query a GO gene set collection and request two outputs:
A single membership flag for the GO term GO:0006915 (renamed to apoptosis). If the mapped gene is in that set, the output cell is yes (otherwise empty).
in_sets, a list of all set IDs (GO terms) that the gene belongs to in the collection.
Note that in this pipeline, gene_list is not marked with internal: false, so it does not appear
in the output. It is still produced and used as input to downstream annotators.
Create a file called annotation_pipeline_5.yaml with the following content:
- effect_annotator:
gene_models: hg38/gene_models/MANE/1.5
genome: hg38/genomes/GRCh38-hg38
attributes:
- genes
- worst_effect
- gene_list # >----->----->----->----->----->----->----->----->----┐
# |
- gene_set_annotator: # |
resource_id: gene_properties/gene_sets/GO_2025-07-22_release. # |
input_gene_list: gene_list # <-----<-----<-----<-----<-----<-----<-----<┘
attributes:
- source: "GO:0006915"
name: apoptosis
- in_sets
To annotate with this pipeline, run:
annotate_columns variants.txt annotation_pipeline_5.yaml -o annotation_5.txt
This produces annotation_5.txt with the effect outputs plus the requested gene set annotations: