GAIn Python interface
The command-line workflow is designed for specific GAIn tasks, such as annotating variants, positions, or regions and inspecting resources in a GRR. This covers many common annotation use cases. However, users may also want to ask more open-ended questions, such as summarizing the contents of a repository, querying a resource over a custom interval, combining information from several resources, or generating a custom plot. The Python interface supports these workflows by allowing users to access GRR resources directly and process the results using standard Python tools.
The first step is to connect to a Genomic Resource Repository (GRR). In the simplest case, a repository can be accessed without providing any arguments:
from gain.genomic_resources.repository_factory import build_genomic_resource_repository
grr = build_genomic_resource_repository()
Calling build_genomic_resource_repository() without arguments uses the default GRR configuration available
in the user environment. In a standard GAIn installation, the default configuration provides access to
the public IossifovLab GRR. Users can also access local repositories or other public repositories by
providing the repository definition explicitly. For example, the code below accesses the current
working directory as a local GRR:
import os
from gain.genomic_resources.repository_factory import build_genomic_resource_repository
grr = build_genomic_resource_repository({
"id": "local_grr",
"type": "directory",
"directory": os.getcwd(),
})
After connecting to a repository, individual resources can be accessed using their resource IDs. For example, users can access a reference genome, a gene models resource, or a position score resource, and then call resource-specific methods on the resulting Python objects. This allows GRR resources to be queried directly in Python for custom analyses that are not part of a predefined annotation workflow.
The examples below illustrate three uses of the Python interface: inspecting chromosome lengths from a reference genome, locating a gene and retrieving scores across its interval, and summarizing the number of resources available for different genome builds. GAIn development page provides more detail on the Python methods available for different resource types.
1: Chromosome lengths
The following example shows how to access a reference genome and retrieve chromosome lengths for a selected set of canonical chromosomes (chromosomes 1-22, X, Y, and M). First, the default GRR is accessed and a reference genome resource is retrieved using its resource ID. The script then defines a set of canonical chromosome names and iterates over the chromosomes available in the genome. For each chromosome that matches the canonical set, the chromosome length is retrieved and printed.
from gain.genomic_resources.repository_factory import build_genomic_resource_repository
grr = build_genomic_resource_repository()
from gain.genomic_resources.reference_genome import build_reference_genome_from_resource_id
genome = build_reference_genome_from_resource_id("hg38/genomes/GRCh38-hg38", grr).open()
# Define canonical chromosomes
canonical_chroms = {f"chr{i}" for i in range(1, 23)}
canonical_chroms.update(["chrX", "chrY", "chrM"])
for chrom in genome.chromosomes:
if chrom in canonical_chroms:
print(chrom, genome.get_chrom_length(chrom))
Save this file as python_1.py, and run it with:
python python_1.py
The output lists the canonical chromosome names and their lengths for the hg38/genomes/GRCh38-hg38 genome
resource. The same chromosome lengths are reported in the
HTML summary page for this resource,
allowing users to compare the Python output with the GAIn resource summary.
chr1 248956422
chr2 242193529
chr3 198295559
chr4 190214555
chr5 181538259
chr6 170805979
chr7 159345973
chr8 145138636
chr9 138394717
chr10 133797422
chr11 135086622
chr12 133275309
chr13 114364328
chr14 107043718
chr15 101991189
chr16 90338345
chr17 83257441
chr18 80373285
chr19 58617616
chr20 64444167
chr21 46709983
chr22 50818468
chrX 156040895
chrY 57227415
chrM 16569
2: Position scores across a gene
This example shows how to retrieve and visualize position scores across a gene (e.g. TP53).
The script uses the MANE 1.5 gene models resource (hg38/gene_models/MANE/1.5) to obtain
the genomic coordinates of the primary transcript. It then queries the phastCons100way position
score resource (hg38/scores/phastCons100way) across that interval to retrieve conservation scores.
The scores are plotted over the TP53 region, with genomic position on the x-axis and the position
score on the y-axis, and the resulting figure is saved as TP53_phastCons100way.png in the current
working directory. This example illustrates how gene models and position score resources can be
combined to extract and visualize signal over a biologically meaningful region.
GENE_NAME = "TP53"
from gain.genomic_resources.repository_factory import build_genomic_resource_repository
grr = build_genomic_resource_repository()
from gain.genomic_resources.gene_models import build_gene_models_from_resource_id
gene_models = build_gene_models_from_resource_id("hg38/gene_models/MANE/1.5", grr).load()
tx = gene_models.gene_models_by_gene_name(GENE_NAME)[0]
chrom, start, end = tx.chrom, tx.tx[0], tx.tx[1]
print(f"{GENE_NAME} is on {chrom}, from position {start} to {end}.")
from gain.genomic_resources.genomic_scores import build_score_from_resource_id
score = build_score_from_resource_id("hg38/scores/phastCons100way", grr).open()
xs = []
ys = []
for pos_begin, pos_end, values in score.fetch_region(chrom, start, end):
if values is not None:
for p in range(pos_begin, pos_end + 1):
xs.append(p)
ys.append(values[0])
import matplotlib.pyplot as plt
plt.figure(figsize=(10, 4))
plt.plot(xs, ys)
plt.xlabel(f"Position on {chrom}")
plt.ylabel("phastCons100way")
plt.title(f"phastCons100way across {GENE_NAME}")
plt.tight_layout()
plt.savefig(f"{GENE_NAME}_phastCons100way.png", dpi=150)
Save this file as python_2.py, and run it as before:
python python_2.py
This will produce the following image:
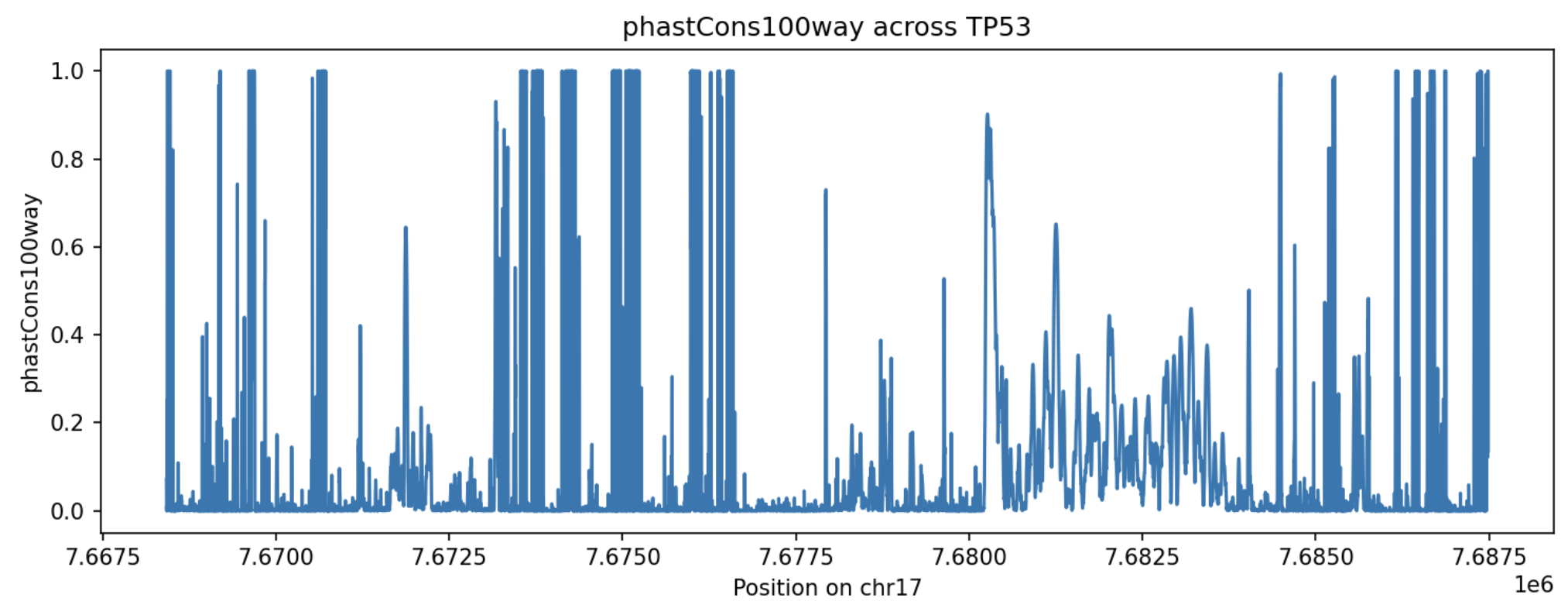
In this plot, higher phastCons100way values indicate positions that are more conserved
across the 100-way vertebrate alignment used by the resource. Peaks in the plot mark parts of
the TP53 interval that are under stronger evolutionary constraint, while lower values indicate
less conserved positions. This provides a compact view of how conservation varies across the TP53 locus.
3: Resource counts by genome
This example summarizes how many resources of selected types are available for each genome build
in the GRR. The script iterates over all resources, assigns each resource to a genome based on
the prefix of its resource ID (e.g., hg19/, hg38/, t2t/), filters by resource type, and counts
how many resources of each type are present for each genome. The result is organized as a table (DataFrame).
import pandas as pd
from collections import defaultdict
from gain.genomic_resources.repository_factory import build_genomic_resource_repository
grr = build_genomic_resource_repository()
genomes = ["hg19", "hg38", "t2t"]
types = ["genome", "gene_models", "position_score", "allele_score", "cnv_collection"]
# initialize counts
counts = {g: defaultdict(int) for g in genomes}
for resource in grr.get_all_resources():
rid = resource.get_id()
rtype = resource.get_type()
for g in genomes:
if rid.startswith(f"{g}/") and rtype in types:
counts[g][rtype] += 1
# build dataframe
df = pd.DataFrame(
{g: [counts[g][t] for t in types] for g in genomes},
index=types
)
df.index.name = "resource_type"
print(df)
Save this file as python_3.py, and run it as before:
python python_3.py
The output is a table with resource types as rows and genome builds as columns, where each entry gives the number of resources of that type for the corresponding genome.
resource_type |
hg19 |
hg38 |
t2t |
|---|---|---|---|
genome |
1 |
3 |
1 |
gene_models |
5 |
47 |
1 |
position_score |
152 |
8 |
0 |
allele_score |
5 |
31 |
0 |
cnv_collection |
0 |
7 |
0 |